Processing Takara Bio ICELL8 single-cell data
Warning
The ICELL8 platform is no longer actively supported within the
auto-process-ngs package; the relevant parts of the
software are deprecated and are likely to be removed in a
future version.
As the code has not been maintained or updated, there is no guarantee that the software will run correctly (or at all), and the documentation may also be inaccurate.
Use of the software for this platform is therefore strongly discouraged and is done at your own risk.
Background
The Takara Bio ICELL8 system prepares single-cell (SC) samples which are then sequenced as part of an Illumina sequencing run.
Two protocols are available:
ICELL8 single cell RNA-seq
Initial processing of the sequencing data produces a single Fastq file R1/R2 pair, where the R1 reads hold an inline barcode and a unique molecular identifier (UMI) for the read pair:
Inline barcode: bases 1-11 in R1
UMI: bases 12-25 in R1
This is illustrated in the figure below:
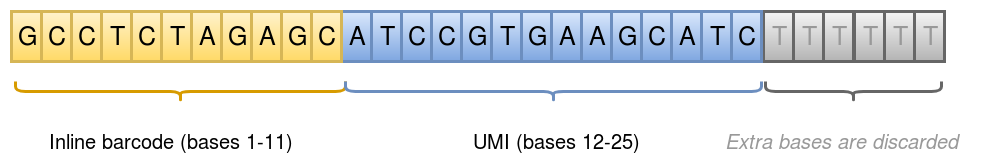
Each inline barcode corresponds to a specific well in the experiment, which in turn corresponds to a specific single cell. The mapping of barcodes to well/cell are given in the Well list file.
The corresponding R2 read contains the actual sequence data corresponding to the cell and UMI referenced by its R1 partner.
Processing protocol for ICELL8 scRNA-seq data
Initial Fastqs can be generated from ICELL8 single-cell8
RNA-seq data using the --protocol=icell8 option of the
make_fastqs command:
auto_process.py make_fastqs --protocol=icell8 ...
Note
--protocol=icell8 runs the standard bcl2fastq commands with
with the following settings:
Disable adapter trimming and masking by setting
--minimum-trimmed-read-length=21and--mask-short-adapter-reads=0(recommended by Wafergen specifically for NextSeq data)Updating the bases mask setting so that only the first 21 bases of the R1 read are kept.
This is recommended to stop unintentional trimming of UMI sequences (which are mostly random) from the R1, should they happen to match part of an adapter sequence.
Once Fastqs have been generated the projects.info file should be
updated with the following values for SC_platform and Library
fields:
Single cell platform |
Library types |
|---|---|
|
|
Running the setup_analysis_dirs command will automatically
transfer these values into the ICELL8 project metadata on creation.
Subsequently the following steps are required:
Run initial QC using the
run_qccommand (thesinglecellQC protocol will be used automatically)Perform ICELL8-specific filtering and additional QC on the reads by running the
process_icell8.pyutility, as described in ICELL8 QC and filtering protocolManually update the sample name information in the
project.infoandREADME.infofiles as described in Appendix: manually updating sample lists
ICELL8 QC and filtering protocol
The process_icell8.py utility script performs initial filtering
and QC according to the protocol described below. The utility also splits
the read pairs into Fastqs by both barcode and by sample.
In addition to the FastqS produced from the make_fastqs-icell8-protocol step, the processing also requires an input Well list file. This lists the valid ICELL8 barcodes and maps those barcodes to their parent sample.
The following steps are performed:
Optional initial quality screen: this applies quality filtering to the barcode and UMI sequences, and rejects read pairs that fail to meet the following criteria:
Barcode bases must have Q >= 10
UMI bases must have Q >= 30
By default this filtering is not used (this is recommended for NextSeq data). The quality filtering can be turned on by specifying the
--quality-filteroption.Barcode filtering: barcodes are checked against the list of expected barcodes in the input “well list” file; read pairs that don’t have an exact match are rejected.
Read trimming:
cutadaptis used to perform the following trimming and filtering operations on the R2 read:
Remove sequencing primers
Remove poly-A/T and poly-N sequences
Apply quality filter of Q <= 25
Remove short reads (<= 20 bases) post-trimming
NB if an R2 read fails any of the filters then the read pair is rejected.
Poly-G region estimation:
cutadaptis used to look for R2 reads which contain poly-G regions. These reads are counted but no other action is taken; the step simply estimates the size of the effect in the data.NB this step is performed after the barcode filtering.
Contamination screen:
fastq_screenis run to check the R2 reads against a set of mammalian and contaminant organisms, and exclude any read pairs where there is an exclusive match to the contaminants.If the screen files aren’t defined in the
auto_process.inifile then they must be explicitly supplied to the utility using the-m/--mammalianand-c/--contaminantsoptions.
Note
The contaminant filtering step can be turned off by specifying
the --no-contaminant-filter option, for example if analysing
data from a non-mammalian organism.
Reorganisation by barcode and sample
At the end of the QC and filter pipeline the read pairs are reorganised in two different ways:
Reorganisation by barcode: the filtered read pairs are sorted into individual Fastqs according to their inline barcodes. This set of Fastqs forms the final outputs of the pipeline. Note that each barcode corresponds to a single cell, and the number of R1/R2 file pairs is equal to the number of barcodes/cells (~1000).
Reorganisation by sample: the read pairs are sorted into Fastqs according to the sample name associated with the barcodes/cells in the “well list” file. Essentially these group all the single cells from each sample, so the number of R1/R2 file pairs corresponds to the number of samples.
The information on valid barcodes and the relationship of barcode to sample are taken from the Well list file.
Each set of Fastqs are stored in their own directories:
fastqs.barcodes and fastqs.samples. Note that the read pairs
themselves are the same in each set.
The standard QC procedure is run on each set of FastqS (barcodes and samples) and QC reports are generated for each.
Outputs and reports
The pipeline directory will contain the following output directories:
Directory
Description and contents
fastqsInitial Fastqs from
bcl2fastq
fastqs.barcodesFastqs with reads sorted by ICELL8 barcode (i.e. cell), plus QC outputs. The Fastqs will be named according to the convention
NAME.BARCODE.r[1|2].fastq.gz.
fastqs.samplesFastqs with reads sorted by ICELL8 sample name (as defined in the input well list file), plus QC outputs. The Fastqs will be named according to the convention
SAMPLE.r[1|2].fastq.gz.
qcQC for the initial Fastqs
qc.barcodesQC for the Fastqs in
fastqs.barcodes
qc.samplesQC for the Fastqs in
fastqs.samples
statsSummary of the read and UMI counts after each processing stage, in TSV (
icell8_stats.tsv) and XLSX format (icell8_stats.xlsx)
logsLogs from the pipeline execution
scriptsScripts generated as part of the pipeline execution.
icell8_processing_dataData and plots for the final summary report (see below)
The directory will also contain:
A copy of the Well list file (name preserved)
A final summary report
icell8_processing.htmlA
README.infofile (nb only if the directory was set up as an autoprocess project)
The final report summarises information on the following:
Numbers of reads assigned to barcodes
Overall numbers of reads filtered after each stage
Initial and final read count distributions against barcodes
Number of reads assigned and filtered at each stage by sample
Poly-G region counts and distribution
ICELL8 single cell ATAC-seq
Warning
This protocol should be considered to be in an “beta” state.
Initial Fastqs can be generated from ICELL8 single-cell8 ATAC-seq data
using the --protocol=icell8_atac option and specifying a
Well list file (via the mandatory --well-list argument):
auto_process.py make_fastqs --protocol=icell8_atac --well-list=WELL_LIST_FILE...
This runs bcl2fastq to perform initial standard demultiplexing based on
the samples defined in the sample sheet, followed by a second round of
demultiplexing into ICELL8 samples based on the contents of the well list
file.
Fastq generation produces a set of R1/R2 and I1/I2 Fastq file pairs for each sample defined in the well list file. The R1 and R2 reads are the actual data for each sample, and the I1 and I2 reads correspond to barcodes in the well list file.
Once Fastqs have been generated the projects.info file should be
updated with the following values for SC_platform and Library
fields:
Single cell platform |
Library types |
|---|---|
|
|
Running the setup_analysis_dirs command will automatically
transfer these values into the ICELL8 project metadata on creation
and the run_qc command can be used to generate the QC (the
ICELL8_scATAC QC protocol will be used automatically).
Well list file
The well list file is a tab-delimited file output from the ICELL8 which amongst other things lists the valid ICELL8 barcodes for the experiment and the mapping of barcodes to samples.
Each barcode corresponds to a well which in turn corresponds to a single cell.
Appendix: configuring the ICELL8 processing pipeline
The running of the pipeline can be configured via command line options,
or by setting the appropriate parameters options in the
auto_process.ini configuration file.
Reference data and quality filtering
Mammalian genome panel:
fastq_screenconf file with the indices for “mammalian” genomes, to use in the contamination filtering step.Set using the
-moption on the command line, or via[icell8] mammalian_conf_filein the configuration file.Contaminant genome panel:
fastq_screenconf file with the indices for “contaminant” genomes, to use in the contamination filtering step.Set using the
-coption on the command line, or via[icell8] contaminant_conf_filein the configuration file.To turn off the contaminant filtering, specify the
--no-contaminant-filteroption.Quality filtering of barcode and UMI sequences: by default read pairs are not removed if the associated barcode or UMI sequences don’t meet the appropriate quality criteria.
To turn on quality filtering, specify the
-q/--quality_filteroption (nb there is no equivalent parameter in the configuration file).
Runtime environment
Environment modules: specify a list of environment modules (separated with commas) to load before running the pipeline.
Set using the
--modulefilesoption on the command line, or via[modulefiles] process_icell8in the configuration file.Job runners and processors: specify job runners and number of processors to use for specific classes of tasks in the pipeline. See Job runners and processors for more details.
Aligner: explicitly specify the aligner (currently either
bowtieorbowtie2) to use for contamination filtering.Set using the
-aoption on the command line, or via[icell8] alignerin the configuration file. (NB if this is not set then an appropriate aligner will be selected automatically from those available in the execution environment.)
Fastq batching
Read batch size: number of reads to assign to each “batch” when splitting Fastqs for processing.
Batching the reads enables many of the pipeline tasks to run in parallel, if the execution environment allows it (e.g. if running on a compute cluster).
Set using the
-soption on the command, or via[icell8] batch_size.
Job control
Maximum number of concurrent jobs: limits the number of processes that the pipeline will attempt to run at any one time.
The default is taken from the
max_concurrent_jobsparameter in the configuration file; it can be set at run time using the-j/--max-jobscommand line option.
Job runners and processors
Job runners and numbers of processors can be explicitly defined for different “stages” of the pipeline, where a stage is essentially a class of tasks).
For the ICell8 processing pipeline the stages are:
Name
Description
contaminant_filter
Tasks for filtering “contaminated” reads
qc
Tasks for performing QC on the Fastqs
statistics
Tasks for generating statistics
Use the -n/--nprocessors and -r/--runners options
to specify the number of cores that can be used, and an appropriate
runner (if necessary) for each of these stages.
Via the command line e.g.:
process_icell.py ... -r statistics='GEJobRunner(-pe smp.pe 4)' -n 4
Via the configuration file:
[icell8]
nprocessors_statistics = 4
[runners]
icell8_statistics = GEJobRunner(-pe smp.pe 4)
Appendix: manually updating sample lists
Currently the processing pipeline implemented in process_icell8.py
doesn’t automatically update the sample lists in projects.info
and the README.info in the ICELL8 project directories.
To do this manually requires extracting the sample names and editing the files to update them with the correct data.
The sample names can be extracted from the well list file using the command:
tail -n +2 WellList.TXT | cut -f5 | sed 's/ /_/g' | sort -u | paste -s -d","
which produces a list suitable for projects.info (e.g.
Pos_Ctrl,SC004,SC005,SC006).
To get them in a format suitable for the README.info file:
tail -n +2 WellList.TXT | cut -f5 | sed 's/ /_/g' | sort -u | paste -s -d"," | sed 's/,/, /g'
(e.g. Pos_Ctrl, SC004, SC005, SC006, SC007).
The number of samples can be obtained by:
tail -n +2 WellList.TXT | cut -f5 | sort -u | wc -l